Welcome to the GMM repository.
SPECIAL ATTRIBUTION: We would like to thank WEHI Senior Research Officer Marek Cmero for developing the FACS merge logic used for this project. The code can be found under the
scriptsdirectory inoperations.py, which was derived from hiscelseq-sample-sheet-generatorrepository.
This codebase contains all assets that will be deployed onto WEHI's Milton HPC via R Shiny. For details on the goals and implementation of this project visit the GMM wiki page.
This markdown file contains the following contents:
server.R: The entry point to this applicationsetup.R: Used for initialisating this applicationui.R: Provides the frontend interfaceR: A sub-directory that contains R componentsscripts: Python files that handle merge logic (developed by WEHI Senior Research Officer, Marek Cmero)data.zip: Contains the sample data to demo the applicationmarkdown_assets: Stores images used for markdown files and the GMM wiki
The VIDEO below provides detailed instructions for setting up and running this application on MacOS/Linux. This Shiny R application is designed to assist in the processing and analysis of genomics metadata through an interactive interface.
Note: For Windows users, manual configuration of Python virtual environments and R dependencies is required due to package conflicts. Unfortunately, we do not have an automated solution for Windows at this time.
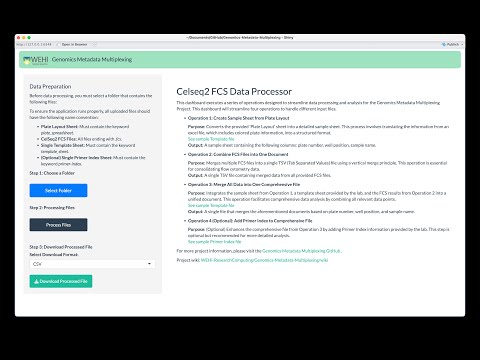
Before proceeding, ensure you have the following prerequisites installed on your system:
- Git: For cloning the repository.
- R: The application is built in R, so ensure you have R installed.
- RStudio (Optional): For a more user-friendly experience running the app locally.
To run the application locally, you can use RStudio or the R console. Here are the steps for both methods:
- Open RStudio: Start RStudio on your local machine.
- Open the
app.RScript: Go toFile>Open Fileand navigate to the location of yourapp.Rscript. - Run the App: Click the 'Run App' button in the RStudio interface to start the application.
-
Start the R Console: Open the R console on your machine.
-
Run the App: Execute the following command in the R console to start the application:
shiny::runApp()
Alternatively, you can use the terminal to run the application:
-
Navigate to the App Directory: Use the
cdcommand to navigate to the directory containing yourapp.Rscript. -
Run the App: Execute the
app.Rscript using RScript:RScript app.R
If you encounter issues, ensure you have the latest versions of R and RStudio. For permission errors, run RStudio as an administrator or use sudo on Linux/MacOS.
Windows users may need to manually configure environments and dependencies due to package conflicts.
We extend our gratitude to all contributors to the Genomics Metadata Multiplexing project. For a complete list of contributors, please visit the Contributors wiki page.